Introduction
Whether you manufacture a medical device, pharmaceutical, automotive or software product that reaches a human end user, you know that quality is one of your top priorities. This is not only because a bad quality or defective product will impact your image, it is also because there are potentially serious consequences resulting from non-conformities of a product or system. Luckily, there is something that can help us: Deviation Management or CAPA.
According to ICH Q7 (Good Manufacturing Practice Guide for Active Pharmaceutical Ingredients), deviations are defined as “Departure from an approved instruction or established standard“. This means, that at any point your product or system is not performing or operating as you intended, or a certain instruction has not been followed – you would raise a deviation.
Within this process lies much reward: Deviations are a tool for the company to strive for improvement and eliminate non-conformities, resulting in a safer, higher quality product and potentially a reduction in waste.
How To: Deviation Management Stages
A procedure known as a Deviation or Non-Conformance Policy will be created by the company, which will outline what is considered a deviation and what to do if one occurs. Deviation Management generally involves the following steps:

Let’s break them down together:
(1) Identification
Our first step is understanding what happened (all details are important here!) and categorizing the deviation. We categorize according to the risk that this deviation poses to the product’s safety, efficacy or quality. Your company may use the following terms:
– Minor
– Medium
– Major
– Critical
(2) Reporting
If it’s not recorded, it didn’t happen. Be sure to follow your company’s policy on how to report deviations and where to document the details you have found during the identification stage.
Anyone in the company can initiate a deviation report, but it is usually done by the responsible department. The deviation will be reported, up to 1 day after the discovery.
It is necessary to be as clear and accurate as possible in the details provided when reporting a non-conformance to aid the investigation. If the deviation happened in production, the manufacturing process will most likely be halted until the matter is investigated. In addition, any immediate corrective actions will be activated, if and when possible to “extinguish the fire”.
(3) Investigation
Part One: Finding the root cause
We have finished recording the events that transpired and understand the final result. Now the real work begins: determining the root cause of the non-conformance or deviation.
We start by brainstorming, often in a meeting composed of various departments, all the possible causes of the deviation. The idea is to form a creative forum where step by step, the causes of the deviation can be narrowed down to the most probable one.
There are a number of tools that can help us succeed in this sometimes-overwhelming process of deviation management. The relevant Department manager & The QA department, with the full cooperation of relevant department(s), initiate the investigation. This may be done using one or more of the following approaches:
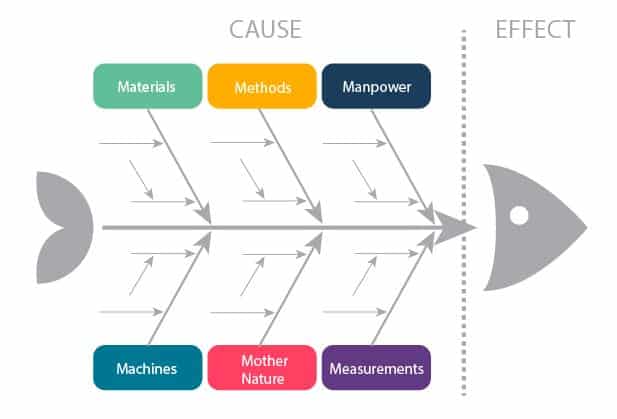
FISHBONE
FISHBONE Analysis (Ishikawa/ Cause and Effect Analysis) – meeting participants agree on a problem statement and then brainstorm all causes within the categories written above.

5W1H
Where, Why, When, Who, What, How

5 WHYs
5 WHYs - a simple technique which can (and should) be incorporated into the approaches used above.
You and your team will determine what is the most appropriate approach for the situation – the central idea in all is to keep asking questions until you find the most probable root cause. When used in conjunction, these approaches are an effective and powerful way to get to the bottom of things!

Part Two: Determining Corrective and Preventative action (s) – CAPA
After analyzing the data gathered in the investigation the most probable root cause(s) are selected and Corrective and Preventive action(s) (CAPA) are determined. These shall be put in place to ensure that this non-conformance and similar non-conformities do not recur.
The CAPA is to be approved by the QA department, but it is the responsibility of the department manager to implement the CAPA and ensure its closure.
(4) Documentation
Following the completion of the investigation process, it is vital that the probable root cause and its corresponding CAPA (s) plan is documented and added to a system where it can be tracked. A due date is to be given for finalizing the activities and a clear plan shall be documented.
(5) Implementation
Time to get to work! We have CAPAs to close!
3 key take-aways
In deviation management:
· Real-time reporting is vital when documenting – we all know that after a few days our memory becomes a little foggy.
· During investigations – use the knowledge and expertise of other departments, because everyone knows their field of work best and can share how to best prevent the next deviation
· Never start an investigation with a prejudice of what is the cause of the non-conformity.
Final Remarks
Deviations are not to be feared – they help the company understand where the problems are and give us the opportunity to fix them. Through corrective and preventative actions (CAPA), we make sure they do not happen again!
Always keep this in mind: Deviation and CAPA are tools for improvement and growth of the company. There are multiple approaches to help you!
What we can suggest
RS NESS is a highly experienced service provider in the Medical Device and Pharmaceutical industries. Our Services include Regulatory Affairs, Clinical Affairs, QA, Validation, and Engineering processes.
We can assist you with Deviation Management and CAPA in helping you to create appropriate policies and process, as well as to optimize deviation handling.
We differentiate ourselves by being quality-oriented. Our technical expertise comes with a hands-on experience and approach, ensuring that our clients receive the most effective and professional service.
Our clients range from small Start-Ups to international companies.
If you have any questions or need professional support, don’t hesitate to get in touch with us.
About the author:

Dganit Haham
QA Project manager
Highly experienced in quality assurance in the Cell Therapy industry. In the past year, Dganit participated in many projects (In the field of Medical Device, FoodTech, CellTherapy, BioPharmceuticals).
Dganit earned her BSc. and MSc. degree in Biochemistry from the Hebrew University of Jerusalem and MBA in BioMed from the Hebrew University of Jerusalem, Israel.




