Risk Management in Medical Device
The EN ISO 14971:2019 standard specifies a process through which we medical device manufacturer can identify hazards associated with the medical device, estimate and evaluate the risks associated with these hazards, control these risks, and monitor the effectiveness of the controls throughout the life cycle of the medical device.
In order to understand the subject, it is essential to understand the terminology:
Hazard– potential source of harm
Hazardous situation- circumstances in which people, property, or the environment are exposed to one or more hazards.
Harm– physical injury or damage to the health of people, or damage to property or the environment.
Now that we know the key definitions, we can understand how they relate to risk management processes. Identify known and foreseeable hazards in normal and fault conditions. It is critical to understand that a hazard can’t result in harm, only hazardous situation. In order to identify how hazard causes a hazardous situation it is important to understand the sequence of initiating events (IV) or circumstances.
|
Hazard |
Initiating Event |
Hazardous Situation |
Harm |
|
Electricity |
Electricity cable wasn’t correctly plugged in |
Exposure to high voltage |
Serious burns/Death |
|
Bioburden |
The sterile barrier package integrity wasn’t examined |
Micro contamination of sterile product |
Bacterial infection |
|
Chemical solution |
Cleaning process wasn’t validated |
Tank contamination with chemical residuals |
Toxic impact |
|
Low temperature |
“Man locked” alarm wasn’t examined in cold room qualification |
Locked employee couldn’t be identified |
Hypothermia |
Let’s take a look at the following table for a number of examples following the sequence in figure 1:
Table 1 Examples of hazards, Initiating events, hazardous situations and harms.
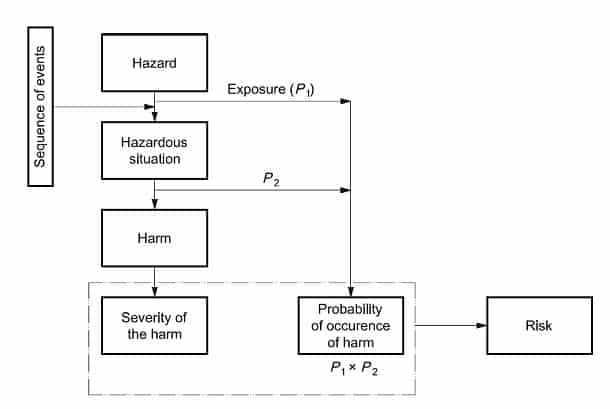
Fig 1: from BS EN ISO 14791 (Annex E)
The common form of the risk priority number (RPN) is a product of the three ratings for severity, occurrence and detection. The detectability can represent the likelihood with which a failure mode is expected to be detected during operation before significant failure effects occur.
Risk management process will be established, implemented, documented and maintained as an ongoing process for:
a) identifying hazards and hazardous situations associated with a medical device
b) estimating and evaluating the associated risks
c) controlling these risks
d) monitoring the
effectiveness of the risk control measures. This process shall include the following elements: risk analysis; risk evaluation; risk control; these elements shall take into consideration production and post-production activities (see Fig 2. Below).
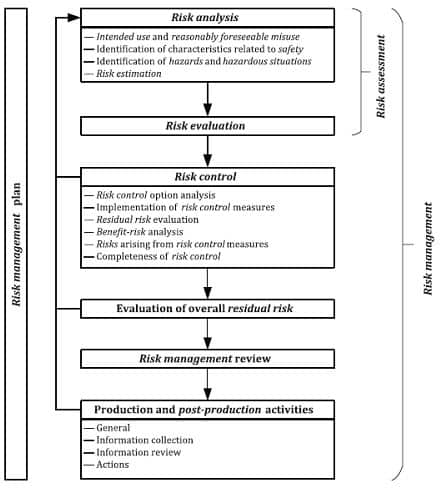
Fig 2: EN ISO 14971:2019 Medical devices – Application of risk management to medical devices
There are a variety of risk analysis techniques including Preliminary Hazard Analysis, Hazard Analysis, Fault Tree Analysis, Failure Mode Effect Analysis and HACCP. These techniques can be complementary, and it might be necessary to use more than one of them.
FMEA – Failure Mode and Effect Analysis (FMEA) is an analytical tool for analyzing potential failures early in the development stage, allowing a manufacturer to identify and act to mitigate a failure.
The purpose of performing FMEA
Identify those failure modes which have unwanted effects on system operation, for example, preclude or significantly degrade operation or affect the safety of the user and other persons Support decisions that reduce the likelihood of failures and their effects, and thus contribute to improved outcomes either directly or through other analyses.
Such improved outcomes include, but are not limited to: Improved reliability Reduced environmental impact Reduced procurement Operating costs and enhanced business reputation.
FMEA can assist with identifying potential causes to a failure and so raise the likelihood of the detection of the failure.
FMEA is a structured approach, and it is usually created within a spreadsheet.
FMEA is created by harnessing the experienced team of specialists from across the company, to observe the development and manufacturing process while identifying failures throughout the known and unknown areas. It can be adapted to meet the needs of any industry or organization, such as: Hardware Software Processes Development Human action and their interfaces, in any combination FMEA can be carried out several times in the lifetime for the same item or process.
preliminary analysis can be conducted during the early stages of design and planning, followed by a more detailed analysis when more information is available. FMEA can include existing controls, or recommended treatments, to reduce the likelihood or the effects of a failure mode. In the case of a closed loop analysis, FMEA allows for evaluation of the effectiveness of any treatment.
All types of FMEA follow the same basic process used for risk assessment and mitigation. For example- System FMEA (also Functional FMEA), Software FMEA, Service FMEA etc.
We will focus on Design FMEA (DFMEA) and Process FMEA (PFMEA), which are the main and most common types of FMEA.
DFMEA- Performed as part of the product design and development stages, in order to identify potential risks in new product or service, or a change in the products design. The identification of risks as early as possible can provide a good opportunity for creating mitigations, even during development stage. Examples for focus points during the performance of DFMEA process can be- componence influence on one another, material used, engineering requirements etc. PFMEA- This process considers all steps involved in a process of producing a component. It is used for identifying risks on process changes as well as in creating a new process. The PFMEA is done under the assumption that the design is adequate, allowing the focus to be on the process. Examples for focus point during the performance of PFMEA can be- machinery, processing method, maintenance, operational requirements etc.
Figure3 is an example of FMEA table.
Fig 3 Extract from an FMEA for an electronic component marking an encapsulation process
References:
- IEC 60812:2018© IEC 2018 – Failure modes and effects analysis (FMEA and FMECA)
- BS EN ISO 14971:2019 – Application of risk management to medical devices
- BS PD CEN ISO/TR 24971:2020 – Medical devices. Guidance on the application of ISO 14971
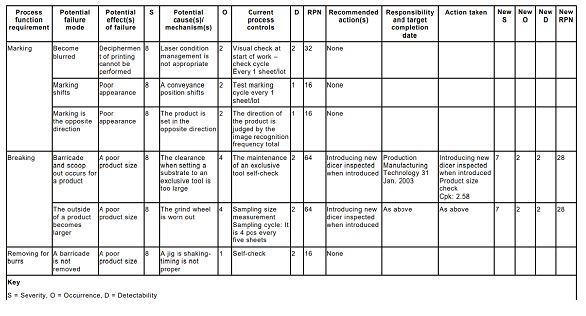
Fig 3 Extract from an FMEA for an electronic component marking and encapsulation process
References:
- IEC 60812:2018© IEC 2018 – Failure modes and effects analysis (FMEA and FMECA)
- BS EN ISO 14971:2019 – Application of risk management to medical devices
- BS PD CEN ISO/TR 24971:2020 – Medical devices. Guidance on the application of ISO 14971




