CHALLENGE
Following an audit conducted in a Medical Device Company, it became apparent that the Device History Record (DHR) release processes are missing as part of the overall manufacturing process. To align with regulatory requirements, the company was required to establish an appropriate Device History Record (DHR) in correlation with the established Design Master Records (DMR).
What is Device History Record (DHR) and why it is so important
Establishment of Device History Records (DHR) falls under 21 CFR Part 820.184. The FDA obligates medical device companies to establish and maintain a device history record (DHR) release process that contains all documentation related to the manufacturing process, quality control (of Raw materials, In-process, and Final Product) packaging and labeling and demonstrates that the device was manufactured according to the approved Device Master Record (DMR) for the specific device.
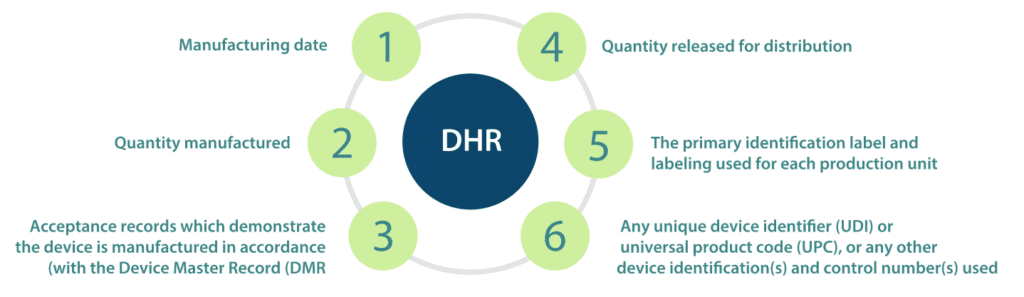
The DHR shall include, or refer to the location of, the following information
- Manufacturing dates;
- Quantity manufactured;
- Quantity released for distribution;
- Acceptance records which demonstrate the device are manufactured in accordance with the Device Master Record (DMR);
- The primary identification label and labelling used for each production unit; and,
- Any unique device identifier (UDI) or universal product code (UPC), or any other device identification(s) and control number(s) used.
In planning product realization, section 7.1 in ISO13485, it states that the organization shall determine the following, as appropriate, required traceability activities specific to the product together with the criteria for product acceptance; a correctly labelled device with assigned identification numbers shall be used for further traceability throughout the supply chain, enabling to identify the root cause in case of any non-conformance and complaints issues related to the product.
The purpose of the DHR release process is to provide evidence that the product was produced according to predefined and validated specifications.
It is a mistake to consider the DHR file as a regulatory requirement only, it is much more than that, as it serves as the most efficient tool and “hard evidence” in assisting you with cases of non-conformances related to the product, for example:
When a complaint is received from the customer, the first thing is to identify the product by the unique identification number (AKA UDI). Your DHR will enable you immediately to know who, under what conditions and when the product was manufactured. In addition, DHR enables us to know which tests were done to the product, and of course the results of those tests. Following the investigation and root cause analysis of the non-conformity, you will be able to know if it can be considered as an isolated event, or a repeatable issue where the company should conduct a corrective and preventive action (CAPA).
The steps of the project
Step 1: Gap assessment
Following gap assessment that was conducted several gaps were identified:
- Missing/lacking documentation
- Lack of compliance with the specifications
- Lack of traceability
- Batch release performed after manufacturing and packaging phases were performed without QA involvement
- Missing control points for critical parameters in the manufacturing process
Step 2: Dedicated and professional team building
Since the DHR involves different disciplines in the organization, such as:
- Manufacturing
- Packaging
- Distribution
- Record Control
- Marketing
- Regulation
- Quality Control
- QA: non-conformity and complaints
The establishment of DHR process release shall involve representatives from all related departments.
Step 3: Manufacturing process deployment
The detailed process was presented initiating from Raw Material receiving through to Final product distribution. All tests are included and the responsibilities are assigned. Also, all documents and records related to the product were collected into a DHR release checklist which resulted in a DHR package ensuring no relevant documentation fell between the cracks or was missed out.
Step 4: Critical parameters identification
The overall manufacturing process was analyzed as part of the preparation work for DHR release process. Once the process is deployed, it is easy to identify where the critical parameters can be tested, and additional acceptance criteria were defined.
In conclusion
The control of the manufacturing process is improved immensely and as a result the company successfully passed the regulatory audits. The main aspects that were improved, are sometimes the very familiar and obvious principles and common practices, however they were not defined as required, or are lacking necessary input. Following are the main points that were improved:
- An appropriate, inclusive and systematic record which covers the DMR and DHR critical points.
- Control points for critical parameters were established and specifications were defined and validated.
- Full traceability of the product was achieved (from Raw Material until Final product)
- QA assumed the responsibility for the DHR release processes.
If you have any questions, or if you need professional support, please contact us.
RS NESS supports the Life Science in such field as: Quality Assurance (QA), Regulatory Affairs (RA), Clinical Affairs (CA), Engineering, Validation and Project Management (PM). We provide service to the companies at different lifecycle stages incorporating end-to-end project activities while adhering to the regulatory requirements. Knowledge, professionalism, and dedication lead our highly qualified team to your success.
About autor

Shani Avneri
Shani has over 17 years experience in the medical device, pharmaceutical, food and cosmetic industry. Shani specializes in process development while integrating quality, regulatory, safety and productivity. Her areas of expertise encompass Quality Control Management, quality support in QMS establishment and application, Audit leading and preparation, Purchasing Control establishment and application, Design Control, Changes and Risk Management.




