Process validation is a formal methodology that allows companies to manufacture products on approved and qualified equipment, with a defined process leading to products that consistently meet their predetermined specifications and quality requirements.
Inadequate Process Validation (PV) is one of the most common issues leadings to warnings from the FDA and other Regulatory Bodies.
The Medical Device and Pharmaceutical industries are very different but share a common goal of bringing safe and effective products to market as quickly and efficiently as possible.
Since there aren’t many PV guidance documents for the medical devices industry in comparison to the pharmaceutical industry, it is not always clear what elements can be adopted in Medical Devices from the Pharmaceutical guidelines.
This paper will outline the main differences between both industries, and what is common between them.
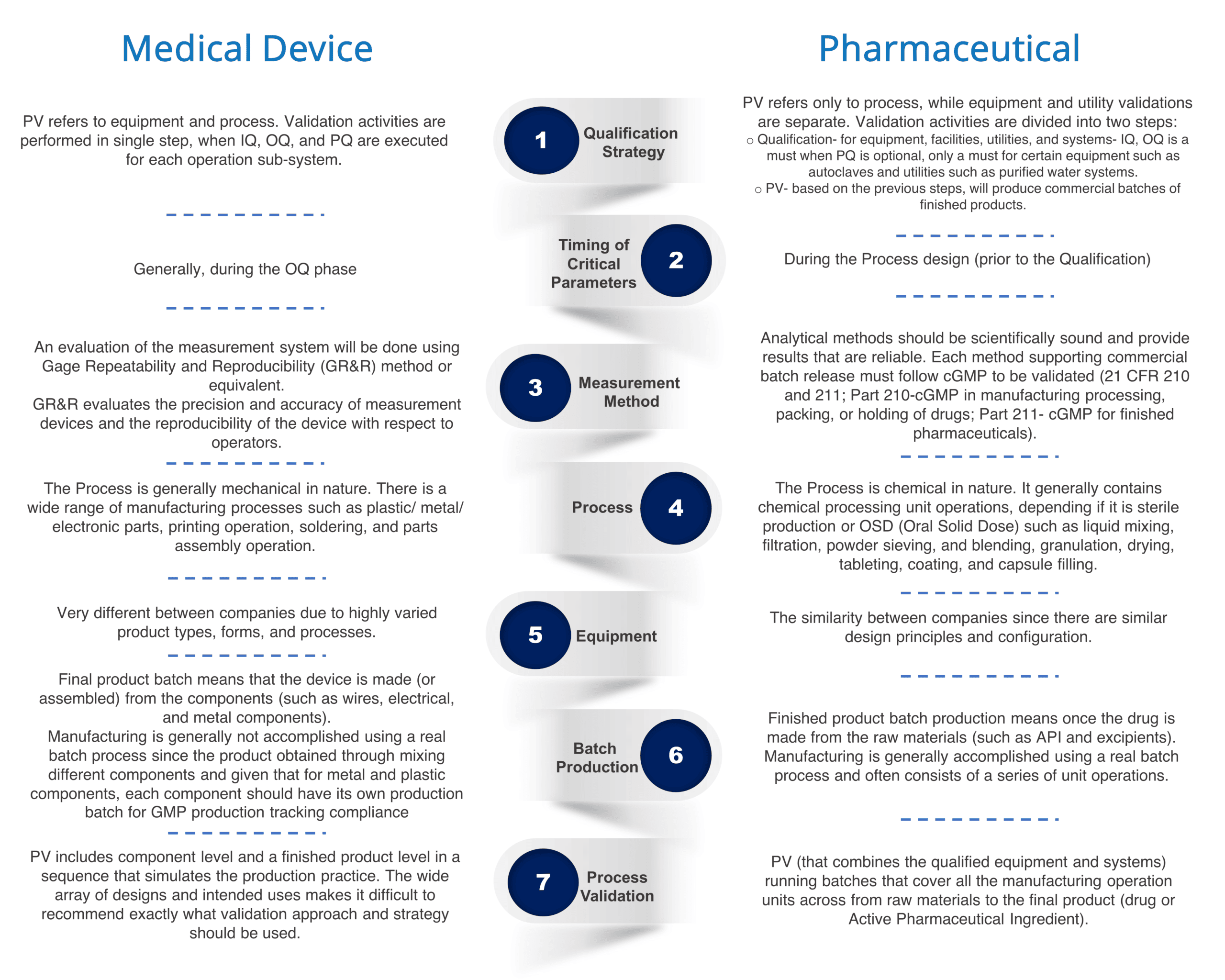
Based on both industries’ main differences, we can notice each industry has a unique basis for initiating PV:
General Strategy
In the medical device industry, the finished product batch is made by the assembly of multiple components, and each component manufacturing has its own manufacturing batch/lot number for traceability.
If each component manufacturing PV follows the pharmaceutical guidance, too many validations will be required for each medical device product.
The medical device industry must develop its own PV Strategy with IQ, OQ, PQ backbone to include the component level and finished product level based on process characteristics.
Process understanding from different production scales
In the pharmaceutical industry, there is a Process Design phase
prior to PV that consists of development and scale-up activities. Laboratory or pilot-scale models designed to be representative of the commercial process can be used to estimate variability for commercial scale. However, the scale-up procedure of a medical device product is executed using Design Control when at various points in the process there are Design Reviews that evaluate the design requirements and make sure the product is on track to meet the specifications. The Design Control requires documentation that details how the medical device product is verified for safety and efficacy. It usually includes User Requirement Specification (URS), Design and Development Planning, Risk Assessment, Design Inputs and Outputs, and Design Verification.
There are also common features between both industries:
Principles for understanding the process
The manufactures in the pharmaceutical and medical device industries should identify and understand the sources of variation possible, detect the presence of each degree of variation, and understand the impact of the variation on the process and product attributes (materials used, process parameters, manufacturing, etc.)
?By what means can we fully understand the process
Risk assessment– this process identifies potential hazards and analyzes what could happen if a hazard occurs. This can be done by several methods such as Failure Mode and Effects Analysis (FMEA).
Engineering studies– examine condition or a set of conditions encompassing upper and lower operating limits and circumstances commonly referred to as “worst-case” conditions.
Continuous Process– Verification (CPV)- in order to enable it, companies should perform, as relevant, extensive controls and monitor process performance and product quality in a timely manner. During the CPV the manufacturing process performance is continuously monitored and evaluated.
Documentation Strategy
Includes Validation Master Plan (VMP) that outline and defines the processes, equipment, and utilities that are to be validated and the priority and order in which this will be done according to Validation Protocols (VPs) and summarized according to Validation Reports (VRs).
Conclusion
Pharmaceuticals and Medical Devices must be manufactured to the highest quality levels. End product testing by itself does not guarantee the quality of the product. Process Validation assists in building the quality into the product and has proven to be an important tool for quality management of Pharmaceuticals and Medical Devices.
RS-NESS provides an umbrella of services to the Life Science industry in the areas of validation, quality assurance, regulatory affairs, clinical affairs, project management, and engineering. This approach enables a “one-stop-shop”, incorporating end-to-end project activities while adhering to the regulatory requirements. Knowledge, professionalism, and dedication lead our highly qualified team to your success.
Key References
- FDA Guidance for Industry, Process Validation: General Principles and Practices (January 2011)
- EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use, Annex 15: Qualification and Validation (March 2015)
- 21 Code of Federal Regulations Parts 210 and 211; Part 210-Current Good Manufacturing Practice in manufacturing processing, packing, or holding of drugs; General Part 211- Current Good Manufacturing Practice for finished pharmaceuticals.
- GHTF/SG3/N99-10, Quality Management Systems – Process Validation Guidance (Edition 2, January 2004)




