:Introduction
In Vitro Diagnostics Directive (IVDD) 98/79/EC will soon be phased out and replaced by the In Vitro Diagnostic Regulation (IVDR) (EU) 2017/746. The IVDR is the new EU legislation applicable to in vitro diagnostic (IVD) medical devices. It entered into force on May 25th, 2017, and is currently in a transition period for manufacturers and economic operators (importers, distributors), and covers all devices that have been regulated under IVDD. The IVDR Date of Application (DOA) is May 2022.
Currently, nearly 90% of In-Vitro Diagnostic (IVD) companies selling products today to the EU under IVDD, have a self-declared CE mark and are minimally overviewed by Notified Bodies, while the main concern is with the products’ technical documentation.
By the end of the transition period, all IVD devices – new and existing – will have to be reclassified and will require Notified Bodies review. Therefore, the vast majority of the previously self-declared industry would require a notified body to certify their products and must undergo extensive changes in the systems and processes, as new classification rules are applied to all CE-marked products.
:IVDR applicability
IVDR Articles 2(2), 2(3) define in vitro diagnostic medical device as “any medical device which is a reagent, reagent product, calibrator, control material, kit, instrument, apparatus, piece of equipment, software or system, whether used alone or in combination, intended by the manufacturer to be used in vitro for the examination of specimens, including blood and tissue donations, derived from the human body, solely or principally for the purpose of providing information on one or more of the following:
(a) concerning a physiological or pathological process or state;
(b) concerning congenital physical or mental impairments;
(c) concerning the predisposition to a medical condition or a disease;
(d) to determine the safety and compatibility with potential recipients;
(e) to predict treatment response or reactions;
(f) to define or monitoring therapeutic measures.”
IVDR also includes Specimen receptacles in its definition: “specimen receptacle means a device, whether of a vacuum-type or not, specifically intended by its manufacturer for the primary containment and preservation of specimens derived from the human body for the purpose of in vitro diagnostic examination;“
?What is an IVD\
?What is NOT an IVD
- Any device intended for a use that examines or monitors humans.
- Any part of a system used to test specimens.
- Accessories for any of the above devices.
- Products for general laboratory use or research-use only products, unless such products, in view of their characteristics, are specifically intended by their manufacturer to be used for in vitro diagnostic examination;
- Invasive sampling devices or those which are directly applied to the human body for the purpose of obtaining a specimen;
- Internationally certified reference materials;
- Materials used for external quality assessment schemes.
:IVDR classification
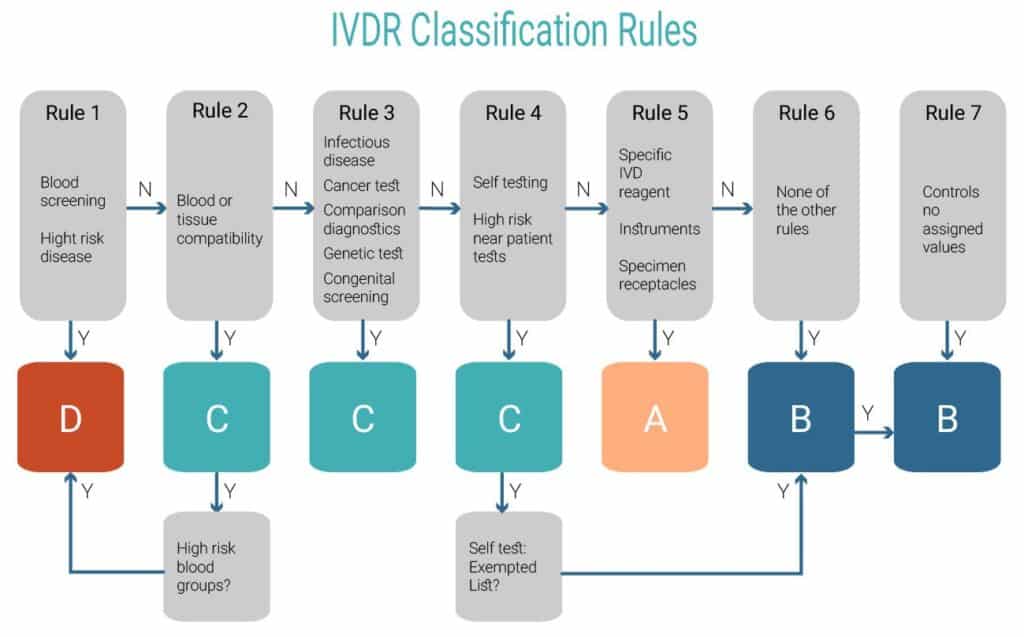
Once you’ve established your device is an IVD under the IVDR, Annex VIII defines the classification rules that you will need for reclassifying your device. This process is mandatory and is the initial “gateway” for downstream activities concerning all aspects of R&D, RA and QA.
IVD classification under IVDR is similar to medical device classification under MDR in the sense that it addresses device classes according to the risk they pose.
The IVDR classification rules include 5 classes of devices: A, A sterile, B, C and D. Class A (non-sterile) manufacturers can remain self-declared, however, all other IVDs A sterile – D require Notified Body approval.
IVD posed risk is measured on two scales: personal risk and public health risk. The risk to public health is considered as the more critical risk. Meaning, that IVDs with even high personal risk may be classified only as high as class C as long as they don’t pose a high public health risk, whereas a mirroring IVD will be the highest class – D.
- Class A sterile – low personal risk and low public health risk;
- Class B – low/moderate personal risk and low public health risk;
- Class C – high personal risk and low/moderate public health risk;
- Class D – high personal risk and high public health risk;
Classification according to Annex VIII goes from the most severe to the least severe of devices: first, you have to eliminate high public health risk (class D), then eliminate the possibility for a high personal risk (class C) and so forth. If your IVD doesn’t fall under any specifically mentioned category – it’s class B.
:Summary
Also note that class A sterile IVDs only require Notified Body approval for the sterile part (the non-sterile is self-declared class A)
The new, rule-based IVDR classification system for devices makes the IVDR more practical, by allowing it to remain relevant to an innovative and growing industry.
As mentioned earlier, The IVDR transition period is scheduled to end very soon, May 25th 2022, and the time is now, sooner than later, for IVD manufacturers to update the technical documentation to comply with the new regulation.
?What we can suggest
RS NESS is a highly experienced service provider in the Medical industry.
RS-Ness can assist you with an IVDR gap assessment to help you create an appropriate project plan, and support the transition throughout the Regulatory, Clinical, QA, Validation, and Engineering processes.
We differentiate ourselves by being quality-oriented and by our technical expertise that comes with a hands-on experience and approach, ensuring that our clients receive the most effective and professional service.
Our clients range from small Start-Ups to international companies.
If you have any questions, or if you need professional support, please contact us.
:About the author

Alina Klepfish
Regulatory Affairs Project Manager
Alina is highly experienced in the Lifescience industry. She participated in and led dozens of projects. She brings with her extensive knowledge in the fields of regulation and project management.
Alina earned a Bachelor of Law degree from the University of Haifa, Israel and a Master of Business Management form the Open University of Israel.




